


Nueva informaciĂłn sobre gen clave para un amplio espectro de enfermedades y caracterĂsticas heredables de humanos
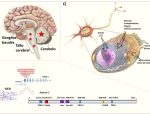
El gen ATAXIN-2 captĂł la atenciĂłn de neurĂłlogos e investigadores de ciencia básica porque se asocia con varias enfermedades neurodegenerativas y con caracteres heredables en humanos Â
Buenos Aires-(Nomyc)-La variación en la longitud del trinucleotido de Citosina Adenina Guanina (CAG), ubicado en el primer exón, causa ataxia e incrementa el riesgo para una decena de enfermedades, desde la Esclerosis Lateral Amiotrófica (ELA), a la de Parkinson o Alzhéimer y Parálisis Progresiva Supranuclear entre otras, enfermedades que son raras o hasta otras más frecuentes, pero  aunque  en muchas solo hay una asociación, sin embargo, la expansión anormal del triplete de CAG sugiere una potencial contribución o modulación al fenotipo resultante.
En esta situación se descubrieron los mecanismos genéticos del gen ATXN2 desde su arquitectura, el gen antisentido ATXN2-AS, la complejidad genética e inestabilidad del triplete de CAG, tanto a nivel intergeneracional como somática y también, se enfatiza en el efecto fundador de la ataxia tipo 2 (SCA2) en Cuba y se presentaron nociones de genética predictiva y se muestran los avances terapéuticos dirigidos a corregir los efectos de la versión defectuosa de este gen.
IntroducciĂłn: ATXN2 pasarĂa desapercibido a los investigadores de no ser por el segmento repetitivo del trinucleĂłtido Citosina Adenina Guanina (CAG) que se encuentra, frecuentemente, interrumpido por tripletes de CAA.
Esta repeticiĂłn mixta de CAG/CAA se encuentra en el primer exĂłn del gen y codifica un motivo repetitivo de glutaminas (poliQ) y el segmento de CAG es inestable y puede llegar a sobrepasar ciertos umbrales en su longitud y con esto causar ataxia espinocerebelosa tipo 2 (aquĂ SCA2) e incrementar el riesgo para otras enfermedades.
De manera habitual, existe un determinismo genĂ©tico donde una mutaciĂłn causa solo una enfermedad con alguna variabilidad o desviaciĂłn del fenotipo más comĂşn, pero sin embargo, la misma mutaciĂłn en ATXN2se asocia con varias enfermedades no tan raras, y a la vez causa un amplio espectro clĂnico en la SCA2.
Variaciones puntuales en la secuencia de ATXN2 dan más de “cincuentatonalidades” a aspectos heredables que van desde el metabolismo de grasas y azucares o nuestra capacidad de discriminar olores, hasta ciertas funciones cerebrales, de ahà que sea un terreno fértil para investigaciones actuales.
En la SCA2 ocurre fundamentalmente una atrofia progresiva del cerebelo y el tallo cerebral, como resultado de la degeneraciĂłn y pĂ©rdida de las neuronas de Purkinje (Fig.1A), lo que causa un sĂndrome cerebeloso caracterizado por la alteraciĂłn de la coordinaciĂłn de los movimientos, ataxia, dismetrĂa o incapacidad para movimientos exactos, incoordinaciĂłn para la ejecuciĂłn de los movimientos de pronosupinaciĂłn de las manos, temblor, marcha inestable, alteraciĂłn del habla y enlentecimiento de los movimientos sacádicos oculares.
El debut de los sĂntomas es de manera general, a los 30 años como promedio, aunque existe anticipaciĂłn genĂ©tica y esta anticipaciĂłn implica que los descendientes portadores podrĂan presentar sĂntomas de la enfermedad a edad más temprana que la del progenitor afectado.
Además, existe una etapa silente o prodrĂłmica que antecede en años al comienzo de la ataxia, donde a pesar del proceso neurodegenerativo no se detectan cambios evidentes del fenotipo cerebeloso, pero sin embargo, hay afectaciones sutiles de la prosodia, la cogniciĂłn, disautonomĂa, y problemas con el balance en la marcha y el equilibrio.
Existen seis interrogantes centrales para este gen ATXN2 de importancia para varias enfermedades y caracteres humanos:
- ÂżCĂłmo se origina la inestabilidad del triplete de CAG de ATXN2?
- ÂżCĂłmo se regula la expresiĂłn del gen ATXN2?
- ¿Cuál es su función fisiológica?
- ÂżCuál es el mecanismo patogĂ©nico de la proteĂna ataxina-2 y sus consecuencias en el fenotipo?
- ÂżEl porquĂ© de la variabilidad fenotĂpica y el pleiotropismo de la mutaciĂłn?
- ÂżCĂłmo cambiar del estado mĂłrbido al saludable?
En este trabajo revió conceptos generales relacionados con cada una de las preguntas anteriores y también proveyó herramientas actualizadas para poder entender la complejidad genética de ATXN2 de importancia para varias enfermedades humanas ya que existe una desproporcionada información acerca de las funciones de ataxina-2 y sus homólogos, pero en este trabajo no pretendemos detallar acerca de esto, si no que más bien trataremos varios temas que no son abordados frecuentemente en la literatura.
Otras enfermedades que origina el mismo Gen: el mecanismo genĂ©tico de expansiones de CAG no es exclusivo de SCA2, existen otras 8 enfermedades neurodegenerativas e incurables causadas por poliQ: enfermedad de Huntington (EH), Ataxias Espinocerebelosas tipo 1, 3, 6, 7, 17, Atrofia Dentado-pálido-rubro-lousiana y Atrofia Muscular Espino-Bulbar que, como rasgo comĂşn en las Ataxias Espinocerebelosas la neurodegeneraciĂłn ocurre en el cerebelo y estructuras relacionadas como las vĂas ascendentes y descendentes de la mĂ©dula espinal, tanto motoras como sensoriales, afectando la esfera motriz, coordinaciĂłn e incluso causando sĂntomas no motrices.
En el caso de la EH la degeneraciĂłn se concentra en un inicio, en el nĂşcleo estriado repercutiendo a nivel motor y del comportamiento, mientras que en la Atrofia Dentado-Pálido-rubro-lousiana se degenera una trĂada de estructuras centrales que incluye nĂşcleos cerebelosos profundos o nĂşcleo dentado, mesencĂ©falo o nĂşcleo rubro o rojo y ganglios de la base o globo pálido, lo que origina un curso desfavorable de la enfermedad con epilepsia, ataxia, movimientos involuntarios, sĂntomas psiquiátricos y demencia.
En la Atrofia Muscular Espino-Bulbar, también conocida como enfermedad de Kennedy, la degeneración de las motoneuronas del tallo cerebral y la espina dorsal resultan en Atrofia y debilitamiento muscular progresivo de curso lento, asà como contracturas.
Todas las enfermedades poliQ, a excepción de la enfermedad de Kennedy, se heredan en base a un patrón autosómico dominante, o lo que es lo mismo, los genes defectuosos residen en cromosomas no sexuales o autosomas y con solo una copia del alelo mutante se desarrollará la enfermedad.
Para las enfermedades poliQ hay una correlaciĂłn inversa entre el tamaño de la expansiĂłn de CAG y la edad de inicio y por otro lado, la enfermedad de Kennedy es recesiva y ligada al cromosoma sexual X, por lo que solo los hombres enfermarĂan al ser XmutY, en tanto que las mujeres XmutX serĂan portadoras asintomáticas de la mutaciĂłn y la gran mayorĂa de las enfermedades poliQ son raras y huĂ©rfanas de tratamiento.
Gen ATXN2 y la proteĂna codificada: el gen fue descubierto al unĂsono en 1996 por tres grupos independientes de EE. UU., Francia y JapĂłn, quienes identificaron la mutaciĂłn causal de SCA2 con metodologĂas distintas: mapeo genĂ©tico, en EE. UU. y uso de anticuerpos monoclonales, en Francia y uso del mĂ©todo de PCR DIRECT, en JapĂłn.
El gen se encuentra en el brazo largo del cromosoma 12 (12q24), se extiende por unas 130kb en el genoma humano y contiene 25 exones con una regiĂłn promotora bipartita que incluye el primer exĂłn y este contiene el segmento repetitivo de CAG y el mismo gen tiene homĂłlogos en otras especies como el ratĂłn, pero en este el motivo poliQ es de solo un residuo (Q).
ATXN2 codifica una proteĂna con funciĂłn, hasta el momento, desconocida y con amplia expresiĂłn en todo el cuerpo humano y en el sistema nervioso tiene una mayor presencia en el cerebelo y la mĂ©dula espinal, mientras que a nivel celular se ha detectado en nĂşcleo, retĂculo endoplasmático rugoso, membrana celular y mitocondria.
Además, está involucrada en el metabolismo del ARN, endocitosis, tráfico del receptor del factor de crecimiento tirosina quinasa, en el metabolismo lipĂdico y de aminoácidos ramificados, en el de la esfingomielina y en el ciclo de Krebs  y las predicciones iniciales acerca de su funciĂłn se obtuvieron del análisis bioinformático de su estructura primaria mostrando varios motivos como PAM, PAM-AD y PR, los cuales la vinculan como una proteĂna de uniĂłn a ARN, lo que está apoyado por hallazgos en modelos de ratĂłn a los que se les ha seleccionado (ATXN2-KO) o insertado (ATXN2-KIN) el motivo poliQ expandido de manera anormal o el gen humano en un cromosoma artificial.
Se conoce además que ATAXINA-2- es responsable del cambio de fase de los gránulos de estrĂ©s, que son cuerpos de ARN y proteĂnas, claves para el metabolismo y regulaciĂłn del ARN que ejercen una funciĂłn global en la regulaciĂłn postraduccional cuando hay estrĂ©s celular.
Sin embargo, la funciĂłn del segmento poliQ aĂşn permanece desconocida, asĂ como los mecanismos patogĂ©nicos derivados despuĂ©s de sobrepasar el umbral patogĂ©nico y estudios de asociaciĂłn recientes señalan que la variaciĂłn del segmento de CAG -o su análogo en la proteĂna -poliQ- estarĂa asociada con la funciĂłn cognitiva, asĂ como con el volumen de varias estructuras cerebrales i.e. tallo cerebral, putamen, globo pálido, tálamo, y amĂgdala.
Esto, a su vez, podrĂa explicar la observaciĂłn de un dĂ©ficit en el aprendizaje visuoespacial en modelos ATXN2-KO asĂ como la existencia de un tercio de individuos atáxicos SCA2 con dĂ©ficit cognitivo, y perdida en la discriminaciĂłn olfativa y por tanto, la perdida crĂłnica de su funciĂłn o su toxicidad pudiera ser clave a lo largo de la vida para el correcto desarrollo cerebral incidiendo en la cogniciĂłn y el aprendizaje.
Nomyc-1-3-21